Ketamine Response Validation Study
Ketamine treatment for depression and other mental illness conditions is fast becoming a life saver for some treatment resistant persons. Recent clinical studies have demonstrated that a single sub-anesthetic dose of ketamine induces rapid and sustained antidepressant actions in treatment-resistant patients.
Unfortunately, only 60% - 70% of patients respond well while 20-30% fail to respond at all. All patients need an initial series of 3-6 infusions over a week to two-week period before they determine if they will respond and to what degree. This initial treatment can cost from $2,000 - $3,000.
For non-responders, this is a costly way to find out they are non-responders.
The solution is to take a genetic test that will cost 10% of the initial treatment and will inform the degree of response to ketamine for individuals.
At Personalized Prescribing Inc., we developed a genetic test that can possibly distinguish between the responders and non-responder based on their unique genetic profiles. However, the test needs to be validated in a clinical setting. We need patients to step up and take the test, either referred by their clinics or their payor organizations. The test is FREE of CHARGE and all participants will be awarded with $100 (CAN) for participating in the study.
1. Overview of the Test
We propose that the periodic effectiveness of ketamine is dependent upon a genetic variability of a set of genes in the Glutamate-GABA excitation inhibition system, NMDA/AMPA receptors degree of inhibition/activation, and ketamine mediated signaling pathways to induce neurogenesis, synaptogenesis, and neuroplasticity.
Personalized Prescribing Inc. (PPI) has developed a genetic panel made up of 28 SNPs in 23 genes that we believe has the capability to determine which patients are likely to respond well to Ketamine. The panel may be able to determine the maintenance frequency of Ketamine injections required for patients that are likely to respond well.
The panel will include genes from the following pathways:
The protein signaling pathway including BDNF, NTRK2, mTOR, GSK3.
The Glutamate receptors, including GRIN2B, GRIN2A, GRIN1, GRIK, and SLC1A2.
The AMPAR expression gene CACNG8.
The GABA pathway, including GABAa and GAD1.
The immune pathway, including IL6, TNFa, IL2b and IL10
The Kynurenine/ Quinolinic pathway, including IDO1, KYAT3, KMO and HAAO
The metabolism of ketamine plays a significant role in ketamine response, so genotyping the CYP2B6, POR and CYP3A4 is included.
2. Patient Process
We will provide participating clinics saliva sample collection kits and consent forms stamped with patient numbers.
Referees (clinics or payor organizations) provide a kit to ELIGIBLE patients. Please note, patient's treatment is not impacted by their participation in the study.
The patient completes the consent form, and if the patient is a known non-responder, they need to complete the ketamine response questionnaire included in the sample collection kit.
The patient follows the instructions for depositing saliva in the collection tube. The patient returns the sample and the completed consent form and ketamine response questionnaire, as applicable by the return envelope provided with the kit.
PPI conducts the genetic test for ketamine response in their own laboratory; the results are uploaded to the Ketamine Response software algorithms.
For patients who didn't start the ketamine infusion, can complete the ketamine response questionnaires after 6 weeks of treatment and send it back to PPI via email at info@personalizedprescribing.com or a research assistant will call the participants to know their response to ketamine treatment.
Patients who complete the questionnaires will receive a $100 Amazon gift card from PPI.
A copy of the "Rx Report- Ketamine Response" report will be sent to patients once the study panel is validated.
3. The Kit
The Kit includes the following:
Saliva Collection Kit
Consent Form
Ketamine Response Questionnaire
Return Envelope
Instruction Card
4. Patient FAQ
Who is eligible to participate?
Patient that tried ketamine treatment and failed
Patients that have decided to commence ketamine treatment
What is required from patients?
A few minutes to complete a short consent form and deposit saliva in a kit that PPI will supply; and patients send both in the return envelop by mail.
What is in it for patients?
They will help others avoid the large cost of determining if ketamine is right for them
They will help science
They will receive an Amazon gift certificate for $100
5. Referee FAQ
What is required from the referee?
Very little; just suggest the participation to eligible patient,
Provide the kit package to patients, the patient will do the rest.
What is in it for referees?
Elevate your clinic to a higher level
Help science
Clinics that participate will receive one free test to provide to their clients when the test is validated, for every patient you refer.
The Science
The Role of Glutamate /GABA in the Cause of Depression
Current research indicates that brain illnesses, including depression, is caused by a dysregulation in one or more “first alert” systems, including the Immune system, the Norepinephrine system, the HPA axis and the Glutamate / GABA system. The dysregulation results in neurodegeneration and disruption of brain plasticity.
In chronic stress and depression:
The HPA axis is inhibited by a GABAergic input from (extra-) hypothalamic and other areas. In chronic stress, GABAergic input is diminished; and/or glutaminergic input from (extra-) hypothalamic sites is increased.
NMDA receptor hyper activation, and or AMPAR inactivation leads to higher amount of Glu remaining in the synapse and inhibiting the activity of GABA and may cause HPA axis dysregulation. An elevated release of glutamate in PFC is repeatedly observed after chronic stress, which is deemed to exert neurotoxicity.
When glutamate release exceeds glutamate uptake, the excess glutamate activates many postsynaptic NMDA-Rs (mostly in GABA neurons), resulting in the induction of excitotoxic neuronal death by allowing excessive Ca2+ influx through the receptor-operated cation channels. Excessive activation of NMDA-Rs and the associated Ca2+ influx result in stimulation of calpain I and nNOS. This causes DNA damage and formation of ONOO─ due to excess NO (nitrosative stress) and other free radicals. The combination of these two changes ultimately leads to mitochondrial dysfunction, cell death and depression.
Ketamine: Mechanism of Action
Ketamine is thought to correct the NMDAR dysfunction above, recent studies propose the following ketamine mechanisms of action as an antidepressant:
1. Disinhibition hypothesis: Ketamine is proposed to selectively block N-methyl-D-aspartate receptors (NMDARs) expressed on GABAergic inhibitory interneurons, which leads to a disinhibition of pyramidal neurons and enhanced glutamatergic firing. Glutamate induced and released binds and activates post-synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) resulting in enhanced brain-derived neurotrophic factor (BDNF) release, activation of the tropomyosin receptor kinase B (TrkB) receptor and subsequently promotion of protein synthesis via the activation of the mechanistic target of rapamycin complex 1 (mTORC1).
2. Inhibition of extra-synaptic NMDARs: Ketamine is proposed to selectively block extra-synaptic GluN2B-containing NMDARs, which are tonically activated by low levels of ambient glutamate regulated by the glutamate transporter 1 located on astrocytes. Inhibition of the extra-synaptic GluN2B-NMDARs is hypothesized to de-suppress mTORC1 function, which in turn will induce protein synthesis.
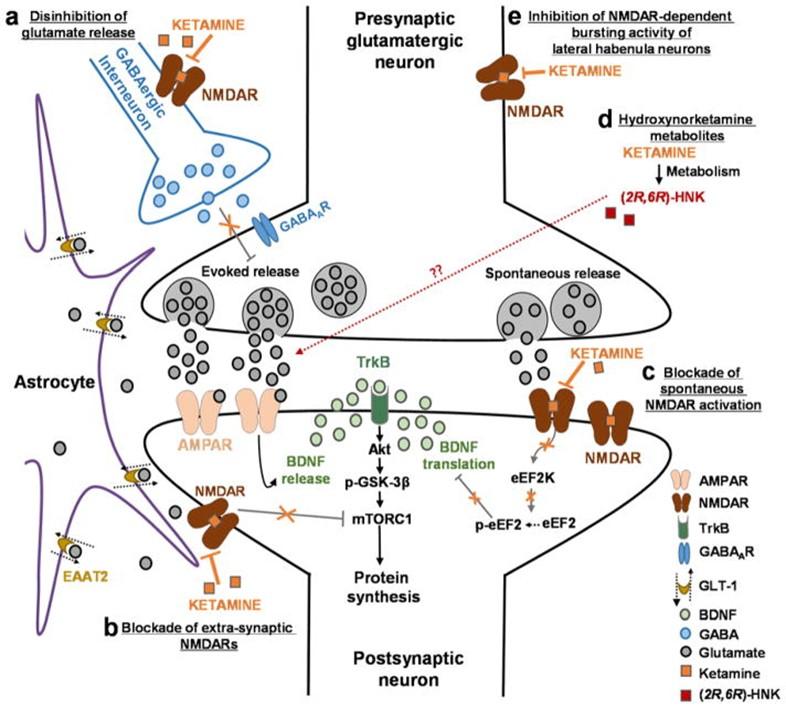
3. Blockade of spontaneous NMDAR activation: Ketamine blocks NMDAR-mediated spontaneous neurotransmission, which results in the inhibition of the eukaryotic elongation factor 2 kinase (eEF2K) activity, thus preventing phosphorylation of its eEF2 substrate. This effect subsequently leads to an enhancement of BDNF translation.
4. Ketamine hydroxynorketamine (HNK) metabolites: Ketamine exerts NMDAR inhibition-independent antidepressant actions via the action of its metabolites, HNKs. Ketamine is metabolized to HNKs following administration, and these HNK metabolites act to promote AMPAR-mediated synaptic potentiation.
These mechanisms of ketamine action are not mutually exclusive and may act complementary in exerting the antidepressant actions of the drug as all hypotheses propose acute changes in synaptic plasticity, leading to sustained strengthening of excitatory synapses, being necessary for antidepressant responses.
Proposed mechanisms of ketamine action as an antidepressant:
(A) Disinhibition hypothesis: Based on the disinhibition hypothesis, ketamine is proposed to selectively block N-methyl-D-aspartate receptors (NMDARs) expressed on GABAergic inhibitory interneurons, which leads to a disinhibition of pyramidal neurons and enhanced glutamatergic firing. Evoked released glutamate binds to and activates post-synaptic α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs) resulting in enhanced brain-derived neurotrophic factor (BDNF) release, activation of the tropomyosin receptor kinase B (TrkB) receptor and subsequently promotion of protein synthesis via the activation of the mechanistic target of rapamycin complex 1 (mTORC1). (B) Inhibition of extra-synaptic NMDARs: Ketamine is proposed to selectively block extra-synaptic GluN2B-containing NMDARs, which are tonically activated by low levels of ambient glutamate regulated by the glutamate transporter 1 located on astrocytes. Inhibition of the extra-synaptic GluN2B-NMDARs is hypothesized to de-suppress mTORC1 function, which in turn will induce protein synthesis. (C) Blockade of spontaneous NMDAR activation: This hypothesis proposes that ketamine blocks NMDAR-mediated spontaneous neurotransmission, which results in the inhibition of the eukaryotic elongation factor 2 kinase (eEF2K) activity, thus preventing phosphorylation of its eEF2 substrate. This effect subsequently leads to an enhancement of BDNF translation. (D) Ketamine hydroxynorketamine (HNK) metabolites: This hypothesis posits that ketamine exerts NMDAR inhibition-independent antidepressant actions via the action of its metabolites, (2R,6R)-HNK and (2S,6S)-HNK. Ketamine is metabolized to HNKs following administration, and these HNK metabolites act to promote AMPAR-mediated synaptic potentiation. These mechanisms of ketamine action are not mutually exclusive and may act complementary in exerting the antidepressant actions of the drug as all hypotheses propose acute changes in synaptic plasticity, leading to sustained strengthening of excitatory synapses, being necessary for antidepressant responses.
1. Mechanism of action including Figure- Mechanisms of Ketamine Action as an Antidepressant, Panos Zanos, Ph.D.a, and Todd D. Gould, M.D., Mol Psychiatry. 2018 April ; 23(4): 801–811.
2. Glutamate and GABA Homeostasis and Neurometabolism in Major Depressive Disorder, Ajay Sarawagi, Narayan Datt Soni and Anant Bahadur Patel, Front. Psychiatry, 27 April 2021
3. Stressor-induced NMDAR dysfunction as a unifying hypothesis for the aetiology, pathogenesis and comorbidity of clinical depression, W.N. Marsden, Medical Hypothesis, Volume 77, Issue 4, October 2011, Pages 508-528